The ongoing pandemic of coronavirus disease 2019 (COVID-19), a respiratory disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has infected billions of people worldwide. Existing studies found that a large proportion of COVID-19 patients had at least one gastrointestinal (GI) symptom1-4, such as diarrhea, vomiting, or belly pain. Importantly, in more than 20% of infected patients, their fecal samples remained positive for the virus even after the respiratory and/or sputum samples exhibited no detectable virus5. In some cases, the viral load in feces is even higher than that in pharyngeal swabs2. All these results suggest that the GI tract might be an important extra-pulmonary site for SARS-CoV-2 infection. Notably, the role of angiotensin-converting enzyme 2 (ACE2) in the invasion of host cells by SARS-CoV-2 via its spike protein is well-established6, and ACE2 is also highly expressed in the small intestine and colon3, 7. Therefore, the prolonged presence of large amounts of fecal SARS-CoV-2 RNA virus is unlikely to be explained by the swallowing of virus particles replicated in the throat but rather suggests enteric infection with SARS-CoV-2.
The human GI tract is the largest immune organ in the body and plays a critical role in the immune response to pathogenic infection or commensal intrusion8. Trillions of microbes live inside the GI tract. Those microbes and their genes, collectively known as the human gut microbiome, modulate host immunity. However, most existing studies on the human microbiome and COVID-19 suffer from the limitations and biases of reference databases and are unable to characterize microbes with high taxonomic resolution. To resolve those issues, we aimed to construct a COVID-19 related metagenomic genome catalog to identify novel taxa and strain-level differences that are likely related to the clinical manifestations of SARS-COV-2 infection.
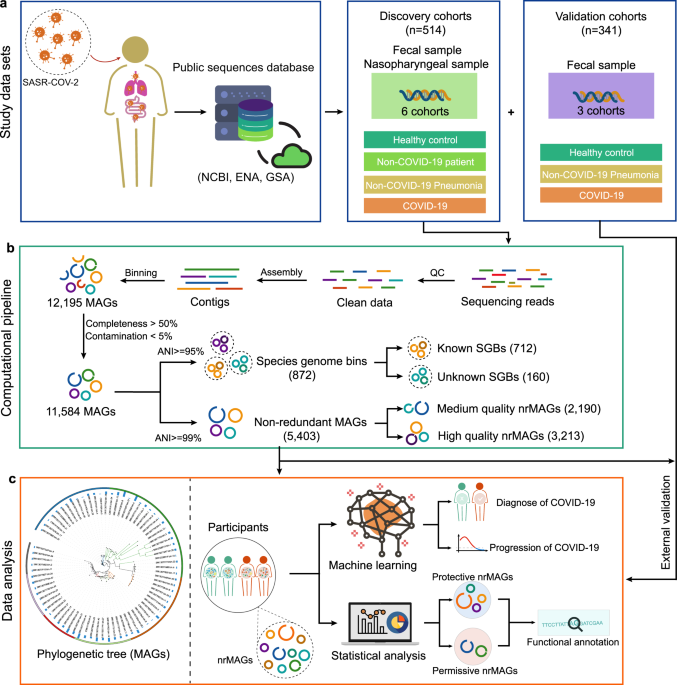
We employed metagenome assembly and binning strategies to reconstruct microbial population genomes directly from the whole metagenome shotgun (WMS) sequencing data of microbiome samples collected from COVID-19 patients and controls. In the discovery cohorts, a total of 514 nasopharyngeal and fecal samples were collected from six independent studies. We recovered a large genome catalog representing 11,584 MAGs and 5,403 Non-redundant MAGs (nrMAGs) of the human microbiome. Through the construction of this microbial genome catalog, we were able to provide the strain-level perspective to dissect the role of the human microbiome in COVID-19. We found for the first time that COVID-19 patients lost many strains (nrMAGs) for certain microbial species when compared to Non-COVID-19 controls in the discovery cohorts. Using machine learning models, we demonstrated that the gut microbiome signatures can accurately distinguish COVID-19 cases from healthy controls and predict the progression of COVID-19.
We then explored the relationship between COVID-19 and the human microbiome at the nrMAGs level. Rather than applying a standard microbe-wide association study (MWAS), we employed the generalized microbe-phenotype triangulation (GMPT)9 method to move beyond the standard association analysis. This novel pipeline enables us to identify a set of COVID-19 related nrMAGs and their determinants (i.e., permissive, and protective) potentially involved in disease pathogenesis. We next investigated whether the functional capacity of permissive and protective nrMAGs differ. Based on the genome annotation, our findings revealed that overrepresentation of permissive nrMAGs and underrepresentation of protective nrMAGs may upregulate the pentose phosphate pathway as their genomes are shown to be highly intact in those relevant modules. In addition to the genome annotation of permissive and protective nrMAGs, we also found that the overall abundance of pentose phosphate pathway in COVID-19 patients was higher than that in the Non-COVID-19 controls. Together, these results suggest that specific microbes may play a role in mediating SRAS-CoV-2 entry into host cells through the pentose phosphate pathway. However, further mechanistic studies are warranted to test the exact role of our candidate permissive and protective nrMAGs in SARS-CoV-2 infection.
To validate our key findings in the discovery cohorts, we collected the raw WMS sequencing data of 341 fecal microbiome samples from three publicly available datasets. Importantly, we demonstrated that the main findings of our study can be largely validated in three independent cohorts.
Overall, our results demonstrate the association between the human microbiome and SARS-COV-2 infection at the strain level and highlight the importance of incorporating the human microbiome in our understanding of SARS-CoV-2 infection and disease progression.
References
- Cheung KS, Hung IFN, Chan PPY, Lung KC, Tso E, Liu R, et al. Gastrointestinal Manifestations of SARS-CoV-2 Infection and Virus Load in Fecal Samples From a Hong Kong Cohort: Systematic Review and Meta-analysis. Gastroenterology 2020; 159:81-95.
- Wolfel R, Corman VM, Guggemos W, Seilmaier M, Zange S, Muller MA, et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020; 581:465-9.
- Liang W, Feng Z, Rao S, Xiao C, Xue X, Lin Z, et al. Diarrhoea may be underestimated: a missing link in 2019 novel coronavirus. Gut 2020; 69:1141-3.
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020; 395:497-506.
- Xiao F, Tang M, Zheng X, Liu Y, Li X, Shan H. Evidence for Gastrointestinal Infection of SARS-CoV-2. Gastroenterology 2020; 158:1831-3 e3.
- Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003; 426:450-4.
- Zhang H, Kang Z, Gong H, Xu D, Wang J, Li Z, et al. Digestive system is a potential route of COVID-19: an analysis of single-cell coexpression pattern of key proteins in viral entry process. Gut 2020; 69:1010-8.
- Chassaing B, Kumar M, Baker MT, Singh V, Vijay-Kumar M. Mammalian gut immunity. Biomed J 2014; 37:246-58.
- Ke S, Xiao Y, Weiss ST, Chen X, Kelly CP, Liu Y-Y. A Computational Method to Dissect Colonization Resistance of the Gut Microbiota against Pathogens. bioRxiv 2022.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in