As a research team, we study cystic fibrosis (CF) – the most common fatal genetic disease in the United States. CF is a monogenic illness, caused by a defect in the gene CFTR. Though CFTR dysfunction has negative effects throughout the body, the primary area of concern is the lungs, where CFTR dysfunction contributes to abnormally sticky, dehydrated airway mucus that impairs airway clearance and encourages colonization by opportunistic pathogens. Over the long term, the result is systemic inflammation, chronic infection, and progressive lung function decline. Though CF comes with a unique list of symptoms, the problems of CF from a microbiological perspective are certainly not unique. The crisis of antimicrobial resistance is one that faces immunocompromised individuals with many different diseases – and work to understand and combat microbial colonization of the CF lung has broad translational implications.
Our team studies CF from a very specific angle – we specialize in microbiology/immunology research, and we also have a keen interest in bioinformatics. We believe that bioinformatics approaches can unlock untapped biological insights hidden in the troves of published data that occupy public databases like NCBI’s Gene Expression Omnibus (GEO). When any group of researchers produces a publication involving a large data set – for example, expression of all bacterial genes after exposure to a specific antibiotic – they are likely to focus on a particular set of genes or biological pathways to tell a concise story in their manuscript. It is entirely fitting that researchers embrace a narrow focus. If they did not, their publications would be far too long and lack clear takeaways to inform other researchers in the field. However, narrowing in on a particular set of genes (as opposed to all genes that are differentially expressed across experimental conditions) does eclipse the discovery of real biological trends in the data that may enhance our understanding of CF biology and host-pathogen interactions.
We believe that it is important to develop software systems that allow researchers to uncover these hidden biological trends in published datasets. To meet this aim, we have designed an R shiny application – called CF-Seq – that allows researchers with diverse scientific and technical backgrounds to interrogate public data from past CF pathogen experiments1.
As the name suggests, the application brings together CF pathogen RNA-sequencing datasets originally published in GEO. In the application, those datasets are characterized by pathogen species and strain – as well as experimental factors like culture medium, experimental treatment, and genes perturbed (either experimentally ‘knocked down’ or ‘over-expressed’). For each dataset, application users can visualize the results of differential gene expression analysis – and explore how the expression of all genes detected in the experiment differ across experimental conditions. They can also explore how broader aspects of biological function (e.g., KEGG biological pathways, GO terms) are impacted by changes in gene expression. The user does not need any computational experience to use CF-Seq, so all CF researchers (and other microbiologists too) can make use of the application to better understand the CF microbiome and even consider new experimental hypotheses to test in the lab.

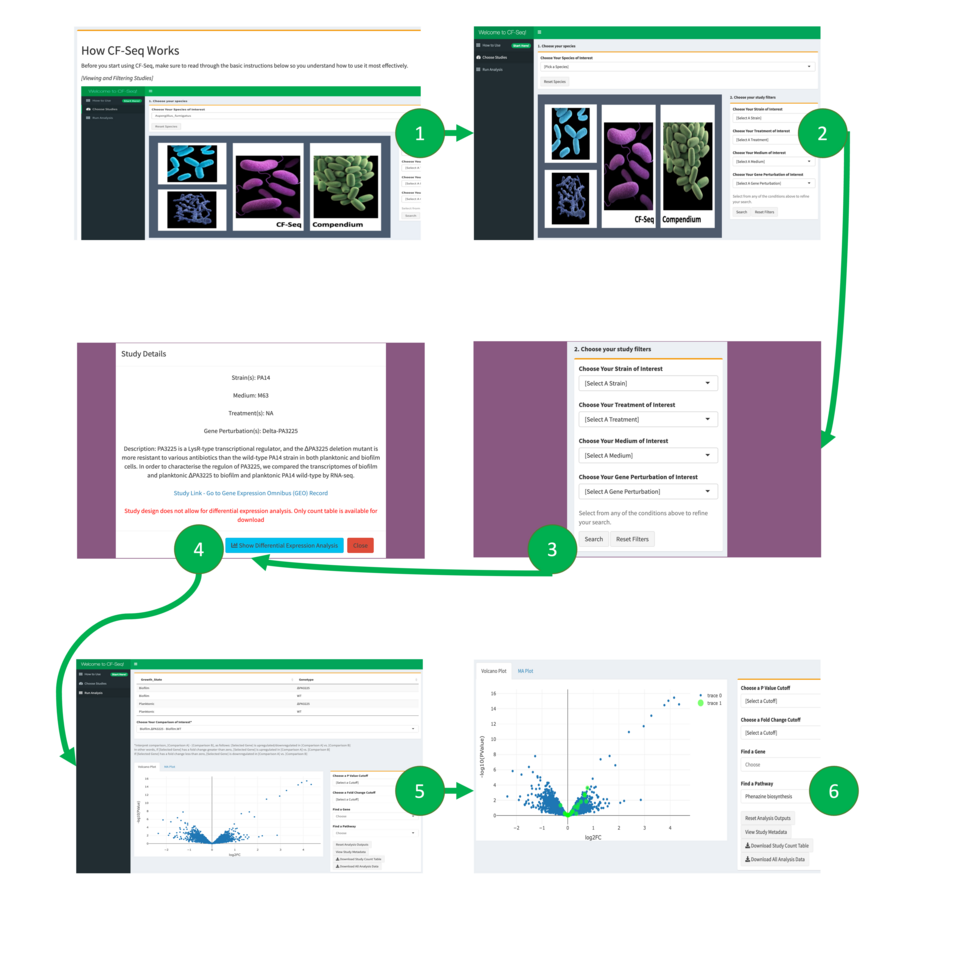
Application workflow for CF-Seq users. Panel 1 shows the starting window of the application, where users are presented with a manual that explains the functionality and purpose of the application. Users are then directed to the study view screen, shown in panel 2, where they can select a species of interest and view available RNA-Seq studies. Panel 3 shows how filters can be applied to delineate studies with certain experimental characteristics (strain, media, treatment, gene perturbed). Panel 4 offers a look at the metadata that can be examined for each individual study. Panels 5 and 6 show the study analysis window, where analysis tables and figures can be generated for all experimental comparisons, individual genes may be highlighted, P value and fold change cutoffs can be selected, and differentially expressed genes on selected KEGG pathways can be highlighted when KEGG pathway information is available (Panel 6). For certain studies, users can also highlight other biological features, such as GO terms, COG categories, and functional descriptions of genes (e.g., “serine/threonine protein kinase”). Zoomed in versions of these figures are available in the supplementary information of our associated publication1.
You might wonder: what specifically can researchers accomplish by re-analyzing and re-interpreting public data? Can the re-use of public data fuel novel projects that merit publication? Just in the realm of CF research (and the related studies on lung / gut biology) alone, researchers are finding creative uses for public datasets that demonstrate their usefulness.
For example, a team of researchers from Emory University recently published a new software tool, built with data from a public repository called Staphopia, to study variation in the quorum sensing system across isolates of common CF pathogen S. aureus. This work offers valuable insights into how bacterial quorum sensing systems evolve over time – and helps lay the groundwork for future surveillance of S. aureus variants of concern2.
Another group of researchers working out of Nanjing Medical University in China have used public data from GEO to show that the percentage of tumor-infiltrating immune cells (and the expression of genes in these immune cells) is correlated with survival in lung cancer patients. This biological information may help guide clinicians in their understanding of disease prognosis – and may even inform new immunotherapy approaches in the long run3.
Also over the past few months, several research teams (University of València, University of Minnesota) have re-analyzed public metagenomic datasets of the human gut microbiome to see how well the data record represents the global population. These efforts are at the head of a data-driven movement to make our knowledge of the human microbiome more representative of the global population and expand metagenomic sequencing initiatives to underrepresented regions of the world4,5.
In all these cases, the re-use of public data for productive, scientific ends is supported by applications like CF-Seq that make data more accessible and show researchers what datasets they have to work with. Over time, we aim to build from CF-Seq – addressing some of the technical limitations discussed in our publication and expanding its reach to more CF pathogen datasets and even CF host gene expression. While our efforts are focused on the CF research space, we hope to promote the CF-Seq application to researchers in other domains – working with different types of public datasets – as an approach to better illuminate the present frontiers of scientific knowledge and spur on future discoveries.
You can access the CF-Seq application online here: http://scangeo.dartmouth.edu/CFSeq/
(And see the source code in our Git Repository: https://github.com/samlo777/cf-seq)
We also invite you to subscribe to our FAIR-CF research newsletter, which aims to uncover all the different ways that researchers are making productive use of public data to drive biological research forward: https://faircf.substack.com/p/fair-cf-spring-2022?r=1hyuwd&s=r&utm_campaign=post&utm_medium=web
References
- Neff, S.L., Hampton, T.H., Puerner, C. et al. CF-Seq, an accessible web application for rapid re-analysis of cystic fibrosis pathogen RNA sequencing studies. Sci Data 9, 343 (2022). https://doi.org/10.1038/s41597-022-01431-1
- Raghuram, V., Alexander, A. M., Loo, H. Q., Petit, R. A., 3rd, Goldberg, J. B., & Read, T. D. Species-Wide Phylogenomics of the Staphylococcus aureus Agr Operon Revealed Convergent Evolution of Frameshift Mutations. Microbiology spectrum, 10(1), e0133421. (2022). https://doi.org/10.1128/spectrum.01334-21
- Wen, S., Peng, W., Chen, Y., Du, X., Xia, J., Shen, B., & Zhou, G. Four differentially expressed genes can predict prognosis and microenvironment immune infiltration in lung cancer: a study based on data from the GEO. BMC cancer, 22(1), 193. (2022). https://doi.org/10.1186/s12885-022-09296-8
- Piquer-Esteban, S., Ruiz-Ruiz, S., Arnau, V., Diaz, W., & Moya, A. Exploring the universal healthy human gut microbiota around the World. Computational and structural biotechnology journal, 20, 421–433. (2021). https://doi.org/10.1016/j.csbj.2021.12.035
- Abdill, R. J., Adamowicz, E. M., & Blekhman, R. Public human microbiome data are dominated by highly developed countries. PLoS biology, 20(2), e3001536. (2022). https://doi.org/10.1371/journal.pbio.3001536

Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in