Why 97%?
Have you ever wondered why 97%? No doubt if you’ve ever sequenced the bacterial 16S rRNA gene, you’ve come across this threshold. But perhaps, like me, you are a little hazy as to its origin?
The mantra that two 16S gene sequences with greater than 95% similarity originate from the same genus, while two with greater than 97% similarity originate from the same species, is persistent – if not always accepted – in current microbiome research.
I found myself wondering about this during our recent re-evaluation of the 16S gene as a tool in microbiome research. A quick literature search led to the work of Stackebrandt and Goebel (1994), and Stackebrandt and Ebers (2006).
Building on previous work to define a DNA reassociation threshold of 70% as a benchmark for bacteria of the same species, these authors plotted the relationship between DNA-DNA reassociation and 16S gene sequence similarity (Figure 1). In both publications, their message was clear: never did they see two bacteria with a 16S gene sequence similarity of <97%, but DNA-DNA reassociation of >70%.

Figure 1: Loosely based on Stackebrandt and Ebers 2006.
There you have it – two bacteria with a 16S gene sequence similarity of <97% are not the same species! We can put this another way – two bacteria that are the same species will have a 16S gene sequence similarity of ≥97%. However, can we state the inverse – that a sequence similarity of ≥97% will mean two bacteria are the same species?
The answer to this last question is unequivocally no. Not only is this poor logic, in both publications mentioned above, the authors provide evidence that bacteria with 16S sequence similarity ≥97% frequently have DNA-DNA reassociation well below the 70% threshold.
This begs the question, if these papers are the source of the 97% threshold, then why has the myth that ≥97% sequence identity constitutes species persisted?
Because it works… until now
One possible reason this inversion fallacy has not been expunged from the microbiome literature is that until recently it has not mattered. It is an open secret that operational taxonomic units (OTUs – generated by clustering sequences with >97% identity) are not a particularly meaningful representation of individual bacterial taxa.
Yet, in spite of their taxonomic vagary OTUs have been genuinely valuable. They are a cornerstone of microbiome research and their use has provided many insights into microbiome community structure. Importantly, though, such insights often come without the need for a detailed understanding of the individual taxa responsible.
Another possible reason why the taxonomic vagary surrounding OTUs has persisted, is that OTUs are not the rate-limiting step when it comes to meaningful taxonomic inference. Sequence length constraints associated with second-generation sequencing platforms restrict the size of the 16S gene region that can be targeted. This limited sequence length in turn limits the ability to distinguish between closely-related bacteria. Put simply, detailed taxonomic resolution is not achievable on current high-throughput short-read sequencing platforms.
What’s changed?
In our recent work we evaluate the potential for high-throughput sequencing of the entire 16S gene to provide species and strain-level taxonomic resolution in microbiome studies. Arguably, the strain is the true functional unit of the microbiome and strain-level quantification should now be the goal of all microbiome researchers. The advent of long-read sequencing technologies, such as PacBio’s circular consensus sequencing (CCS), brings this goal one step closer.
Using CCS, our work highlights two other open secrets in the microbiome field. First, the fact that many bacterial genomes contain multiple 16S gene copies and, second, that these 16S gene copies may diverge with respect to their sequence content. This first issue has historically confounded attempts to calculate relative abundance of bacteria with different 16S copy numbers. The second presents a thorny problem for modern denoising approaches that advocate treating 16S sequences diverging by as little as one base as discrete taxonomic units.

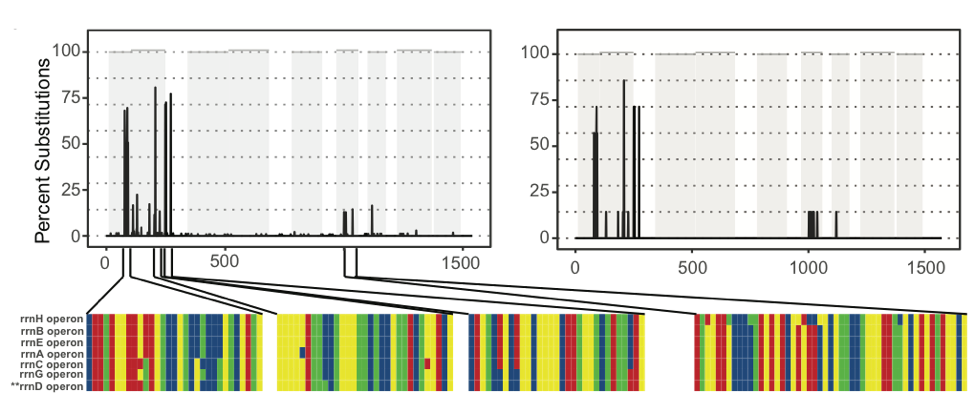
Figure 2: Observed (left) and predicted (right) nucleotide substitution profiles, based on aligning PacBio CCS sequences (left) or reference sequences (right) for the seven 16S genes in the Escherichia coli K-12 substr. MG1655 genome.
Our paper argues that, rather than being problems, these characteristics of the microbiome could instead be leveraged to boost taxonomic resolution. Intra-genomic variation in 16S gene copies creates distinctive substitution profiles when aligning all the 16S genes within a genome (Figure 2). Inter-genomic variation at one multiple homologous loci therefore has the potential to help discriminate between closely-related bacterial taxa.
Our conclusion is that properly accounting for both inter- and intra-genomic 16S gene copy variation presents intriguing new opportunities for the 16S gene as a tool in modern microbiome research.


Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in
Dear Jethro,
Woo, That was very interesting. I personally, have given up using OTU clustering at all. Thank you for your blog.
I favour oligotyping above OTU clustering. I think oligotyping is an underused strategy but it has been working very well for my research in the last few years.
I am also confident that full-length bacterial 16S sequencing together with oligotyping has strong potential to go far beyond genus classification. At least for species level. I don't have experience with strain classification using 16S but it might be promising,
Thanks for your contribution.
Josef